Running NRLMOL excited state calculations¶
Introduction¶
Running excited state calculations is a little complicated at first, this guide will show you how to setup an excited state run. The overall steps required for this are:
- Adding the excited state subroutines to the source code.
- Modfying the Makefile.
- Uncommenting the calling subroutine.
- Setting up a directory to run an excited state.
- Copying files to the directory.
- Modifying the RUNS and SYMBOL files when necessary.
- Creating the OCCEXC file.
The excited state subroutines are not directly a part of the NRLMOL source code, they are kept separate. So, some modifications to the way the NRLMOL binary (or executable) is built must be done. They must be added to the directory were the rest of the source code is located, the file that compiles all the files together must be modified to add these subroutines and the call to the subroutines in the source code must be habilitated.
The subroutines implement the pertubative \(\Delta\)-SCF calculation, developed by Baruah and Pederson (see bibliography for a full description).
Installation¶
In order to run an excited state calculation, first of all you need the excited state subroutines for NRLMOL. Go to the directory where you have your source code and look for these files.
- exc_serial.f
- excrewrite.f
- fill_global.F90
- fill_local.F90
- init_mpi.F90
- orthogonalize_hole.F90
- lowden.f
- particles.f
If you do not have them, then you need to contact your code support group to get them. Put this files in that same directory. Next, you have to modify the Makefile so that they are compiled along with the rest of the source files and become part of the binary nrlmol_exe file. Open the file called Makefile, there should be a section called EXCITED that contains these files:
EXCITED = lowden.o particles.o excrewrite.o ...
These targets are not compiled and built into the final binary file. They must be included as part of the obehct files to be compiled and linked. Add this file group to the OBJ group.
OBJ = $(FTNBASICS) $(FBASICS) $(EXTRAFIL) $(POSTACTIVE) $(DISP) $(BASIS) $(GIT) $(WFX) $(LEVY_PERDEW) $(PCMOBJ) $(EFPOBJ) $(EXCITED)
Next, open the file main.ftn in the source code directory and look for this part of the code:
CALL CHECK_INPUTS
SELECT CASE (ISTSCF)
CASE (4)
CALL READWF(FAILED)
IF (FAILED) THEN
ISTSCF=3
CALL NEWWAVE_SERIAL(ITTOT,TRACE)
END IF
GO TO 111
CASE (11)
CALL READWF2(FAILED)
IF(FAILED) THEN
WRITE(6,*)'ERROR:COULD NOT CREATE SPIN POL FROM SPIN UNPOL'
CALL STOPIT
ENDIF
ISTSCF=4
GO TO 111
END SELECT
IF (EXCITED1) THEN
C CALL EXCWAVE(EXCCONV,TRACE)
ELSE
CALL NEWWAVE_SERIAL(ITTOT,TRACE)
END IF
In this case, we need to uncomment the call to EXCWAVE. That is, we need to remove the C at the beggining of the line that comes after the line IF (EXCITED1) THEN. All you have to do now is recompile the code (you may need to change parameters but you’ll know that until you run the code, so just go ahead and do:
make
NOTE.- You may already have lowden.f in the soruce code since it is used for another type of calculation (fragment analysis) but it’s always better to update it, since source files are constantly under maintenance.
Ground state calculation¶
To run an excited state calculation, a spin polarized ground state calculation of an optimized structure needs to be executed first. You can start this ground state calculation from the CLUSTER file, or generate it from the last geometry printed (in the file XMOL.DAT) when the structure was optimized. The only requirement here is that in the NRLMOL_INPUT.DAT you must set the switch CALC_TYPE to SCF-ONLY. This prevents the program from creating other geometries in SYMBOL.
Open the file NRLMOL_INPUT.DAT and change the following line:
CALCTYPEV = ‘SCF-ONLY’
In the case you forgot to set the CALC_TYPE switch when you submitted the ground state calculation, or just have a previously run ground state calculation.
In the file SYMBOL, you must remove other geometries that may have been created, and correct the number of electrons.
Open the file and scroll down until you find the line:
1 CALCULATION SETUP BY ISETUP
This is the first line of the first geometry, it ends at the line:
EXTRABASIS = 0
If any ohter geometries were created during the ground state calcultation, they will begin after the first geometry this with:
N NEW CALCULATION CREATED BY UPDATE
Where N is the number of geometry, they also end with the EXTRABASIS line. Each geometry also ending with the line:
EXTRABASIS = 0
So remove them all from SYMBOL and just keep the first geometry.
To correct the number of electrons
Look for the line before the EXTRABASIS line just descibed previously, it should have the form:
ELECTRONS = 18.500000 17.500000
Where the numbers should add up to the total number of electrons in you system. You need to make both numbers whole by transfering the partial charge of one of the number to the other one, and make the second number negative. The correct entry fro the above example would be:
ELECTRONS = 18.000000 -18.000000
In the case that you ran a spin unpolarized ground state calculation, you can read the section on how you can turn a spin unpolarized calculation into a spin polarized calculation.
Setup¶
As we already stated, to perform an excited state calculation you must have already performed a spin polarized ground state calculation. Also, never run an excited state calculation in the same directory where you ran the ground state calculation, it’s preferrable that you make a subdirectory in the directory where you ran the ground state calcutation. Once that is done, copy these files from the ground state cacluation to the directory where you will perform the excited state calculation:
- RUNS
- GRPMAT
- REPMAT
- SYMBOL
- ISYMGEN
- HAMOLD
- VMOLD
- NRLMOL_INPUT.DAT
- XMOL.DAT
- A job script file
Also, if you have have used pseudo-potential in any atom of your structure (that is, specified BHS instead of ALL in the CLUSTER file) you will need to copy the file PSPINP as well.
There is an extra file required called OCCEXC, but it will generated separately (discussed in a later section).
Three of the previously listed files need to be modified for the calculation to work properly.
- The NRLMOL_INPUT.DAT file must contain the requirement to do an excited state:
EXCITEDV = ‘Y’ ! Determines if this is an excited state calculation
- The file RUNS must contain this:
-1 1 ITBEG, NCALC
1 4 START: 0=SCR.NUC, 1=HAM, 2=POT, 3=LSF, 4=WFUNC
0 START HAMILTONIAN IS INTERPOLATED: 0=NO, 1=YES
The first number in the first line (-1) tells the program use the existing integration mesh (thus saving some time), the second number (1) tells the program to start from the first geometry listed in SYMBOL. In the second line, the first number (1) tells the program to start from the Hamiltonian matrix (stored in HAMOLD).
- Your SYMBOL file must contain ony one geometry, verify that it does. If not, remove any other geometries on it.
Understanding orbital levels¶
Before we continue, it’s good to cover a review of how the orbital levels of an atom or molecule are managed by NRLMOL.
Remember that we are analyzing the energy levels that electrons occupy for the given structure or atom. These levels are occupied from the lowest energy up to a given level wich is called the highest occupued molecular orbital level (HOMO for short). Above it, all the levels are unoccupied. Of these, our reference is the lowest unoccupied molecular orbital level (LUMO). Between them, the Fermi energy is the level that separates the HOMO and LUMO. This is shown in the figure.

In NRLMOL, the occupancy is displayed in a file called EVALUES.
You need to scroll down until you see the line that starts with FERMI LEVEL, which will contain the Fermi energy. After this, the next line says:SUMMARY OF EVALUES AND THEIR OCCUPANCIES:. From here, there is a listing of the energy levels, however, they are upside down as what is shown in the previous figure. The first level shown is the lowest energy one, and they increase in energy as we scroll down.
A typical line might look like this:
2 REP: 2 DEG: 3 SPIN: 1 ENERGY: -0.347180 OCC: 1.00000
Here, the definitions are:
- The first number is the state number; however this is not excactly true. NRLMOL lists the states incrementally, but remember that since we are dealing with a spin polarized calculation, these levels are for both spins, so they level numbers do not correspond to the actual levels for a given spin.
- The number after REP: is the representation number, this number will be different when the structure contains symmetry.
- The number after DEG: is the degeneracy.
- The number after SPIN: is the spin of that level.
- The number after ENERGY: is the energy for that level.
- The number after OCC: specicies the occupation. This number is 1.00000 when the level is occupied, the last level with this values is the HOMO. Occupation is zero or less than one if it is unoccupied, the first level with this non-one value is the LUMO.
As an exercise, verify that the Fermi level discussed above falls between the HOMO and LUMO levels. Remember, the energy levels are intermixed with the spins, the get the actual number of levels, you would have to manually count them according to their spin. Most of the times the levels are evenly spread (that is, same number of levels for each spin) but this is not always true.
An excited state calculation is calculating the energy to take an electron from an occupied state to an unoccupied state. We sometimes use the language of saying that were are going to move an electron from a HOMO to a LUMO. This is not completely true, since there is only one level that is HOMO and one level that is LUMO. However, we normally call the occupied levels HOMO, HOMO-1, HOM-2 and so forth, and the unoccupied levels LUMO, LUMO+1, LUMO+2, etc.
So when you submit a calculation you might be doing one of a number of possible transitions: HOMO to LUMO (probably being the most common), HOMO to LUMO+2, HOMO-2 to LUMO+2 to name a few.
The OCCEXC file¶
Performing an excited state calculation is calculating the energy required to take and electron from an occupied state (thus creating a hole state) and putting it in an unoccupied state (creating a particle state), this transition is explicitly set in the file OCCEXC and shown in the figure.
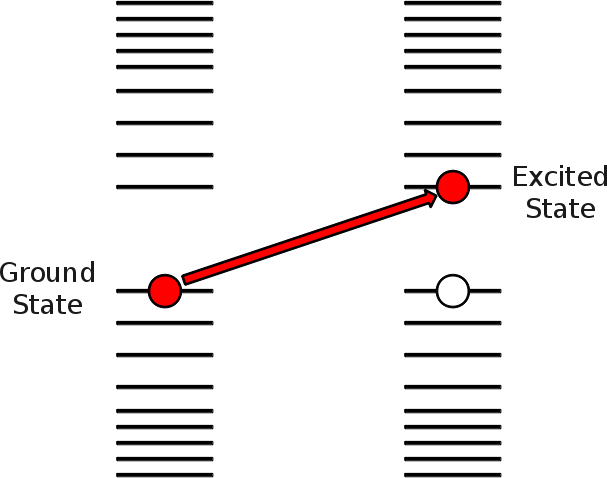
Here is a sample OCCEXC file.
1
-679.225046
1 53 Hole Spin, hole state
2 54 Particle Spin, particle state
53
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1
53
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1
53
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 0
54
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1
- The number in the first line (1) represents the number of transitions we are calculating (you can do more than one transition, but I’ve never done it).
- The number in the second line (-679.226046) is the ground state energy, you can get this value from the ground state calculation in the SUMMARY (the last line in the file) or GEOCNVRG files.
- The next two lines describe the transition to be calculated. The first contains the spin of the level where the hole will be created, and the number of the level where the hole will be created, for this particular case, the hole will be created in the HOMO for spin 1. It’s almost redundant to say that the hole level must be one of the occupied levels.
- The following line describes the spin and energy level where the particle state will be created (the unoccupied state where the electron will jump to). In this case, it can be seen that the electron will change its spin as it goes to an unoccipied level (LUMO in this case). This transition is called a singlet state, when the electron retains its spin, then it is called a triplet state.
- Next, we have a listing of the occupied levels. In this case, we see 53 which is the number of occupied levels, the block after it is 53 number ones, one for each occupied level up to the Fermi level. This information is duplicated for the levels of spin 2. We can consider this as the “before” state previous to the transition. What this is telling us is that there are 53 occupied levevls for each spin. There are of course unoccupied levels, but only the occupuied ones are listed.
- The last two blocks can be seen as the “after” state, when the electron has jumped. For spin one we have the same 53 levels before the Fermi level. However. It can be seen from the listing of number ones that the last one is missing, this means that the HOMO level of spin 1 is now unoccupied. For the block for spin 2, it now lists 54 levels, that’s because there is now an extra eletron for that spin, that is there is now an electron in the first level after HOMO (of course the LUMO).
So we can conclude here that this OCCEXC file describes that we will calculate the transition of HOMO to LUMO in a singlet state.
Once you create this file an put it with the rest of files you are ready to submit your calculation.
Interpreting results¶
Once your calculation has finished a file summary is created, in this case it’s called SUMMARY-EXC, it’s structure is different than the SUMMARY file that the ground state calculation creates, we should note that a new SUMMARY file is created, and it also contains the same information of each iteration but now using values for the excited state calculation.
The first line of the new file SUMMARY-EXC will look something like this:
ALP_OPT: 0.3439 EXCITED ENERGY: -679.225046 3.594751228
- The value listed after ALP_OPT: is the value of \(\alpha\) that was minimized during the perturbative \(\Delta\)-SCF cycle.
- The value after EXCITED ENERGY: is the final ground state energy, the value for the energy in the excited state calculation is the last entry in the SUMMARY file.
- The last value is the transition energy; that is the difference between the ground state and excited state energy, however, it has already been converted from atomic units (Hartree) to electronVolt (multiplied by the conversion factor of 27.2116).
Converting a spin unpolarized calculation to polarized¶
In the case that you ran a spin unpolarized calculation, follow these steps to run a extra iteration and convert your calculation to a spin polarized calculation.
- First of, you may need to recompile the code, since that the value of MXSPN must be set to 2 instead of 1 the PARAMS file. If it’s already set to that, then there’s no need to recompile (but you souldn’t have run a spin unpolarized calculation with this value, it just wastes memory!).
- Open the RUNS file and set it to this:
-1 N ITBEG, NCALC
11 4 START: 0=SCR.NUC, 1=HAM, 2=POT, 3=LSF, 4=WFUNC
0 START HAMILTONIAN IS INTERPOLATED: 0=NO, 1=YES
Where N is the number of the geometry in the SYMBOL file that you want to use.
- Open the SYMBOL file and at the end of the geometry that you will use, look for these lines
ELECTRONS = N M
EXTRABASIS = 0
Where N and M are the electrons of the system, you need to add them and divide them by two (it should be and even number) place this number as positive where N was and place it as negative where M was.
4. Open the NRLMOL_INPUT.DAT file and put Y in the NONSCF option. This will make NRLMOL do a single iteration (where not optimizing the structure), so you don’t have to perform all the iteations in an SCF cycle. Once all of the above has been setup, resubmit your calculation.
NOTE.- This option is a little buggy (we have not tested it throughly), so use with caution.
Bibliography¶
- Tunna Baruah & Mark R. Pederson, DFT Calculations on Charge-Transfer States of a Carotenoid-Porphyrin-C60 Molecular Triad, J. Chem. Theory and Comp. 5, 834 (2009).