Running fragment analysis with NRLMOL¶
This tutorial shows how to perform fragment analysis with NRLMOL code, changes have been introduced in the NRLMOL_INPUT.dat file to request fragment analysis and the structure of the input file CLUSTER has been modified to accomodate these changes. If fragment analysis is not requested, the CLUSTER file remains unchanged.
Getting the code¶
Above all, you will need to obtain the latest NRLMOL version which supports fragment analysis, go to where you have your source code and look for a file called fragment.f90, if it is not present, contact me to update your code.
Steps¶
Individual Fragment Calculation¶
This part of the calculation doesn’t actually require the latest code to be performed. You just need to perform a single NRLMOL run separately on each of the fragments to be analyzed (single point calculation), we do not want to optimize this individual fragments, because the geometry is different when they are taken as a complex. Once the calculation is done, we will need the generated HAMOLD file from each of the fragments. These files must be renamed HAM.1, HAM.2, etc. depending on the number of fragments of the complex. Also, in the file EVALUES we need to collect the following information:
- The number of basis functions: Get the number next to NUMBER OF BASES:
- The number of occupied states: Count the number of occupied states in the list after SUMMARY OF EVALUES AND THEIR OCCUPANCIES: (count the number of states that have OCC: 1)
Complex calculation¶
Now you can perform a regular calculation on the whole complex, but, there are some considerations that must be taken. NRLMOL reorders the atoms you normally specify in the CLUSTER file, So we lose track of what atoms belong to what fragment, for example: if two fragments of a complex contain Hydrogen, NRLMOL will list all Hydrogens together (in generated files like SYMBOL and ISYMGEN) independently of what fragment they belong to. This is problematic, because the hamiltonian matrix is constructed with this sorted order. But we want it to be written so as to reflect the order of the atoms by fragment. So before you submit your compounded CLUSTER file for execution, make shure that it has the atoms in the order that the fragments are speficied, and provide information as to many atoms there are in each fragment.
Fragment analysis¶
A second run of another binary that must be compiled separately from the Makefile performs the actual fragment analysis. Some inputs from the previous individual fragment and complex calculations are required, as well a new input file called FRAGMENT.IN
Example¶
Fragments¶
For an example of a fragment calculation we will use TCNE-Benzene, a small 22 atom complex that can be decomposed into two fragments: a 12 atom benzene ring and the 10 atom TCNE. The complete complex is shown in the figure
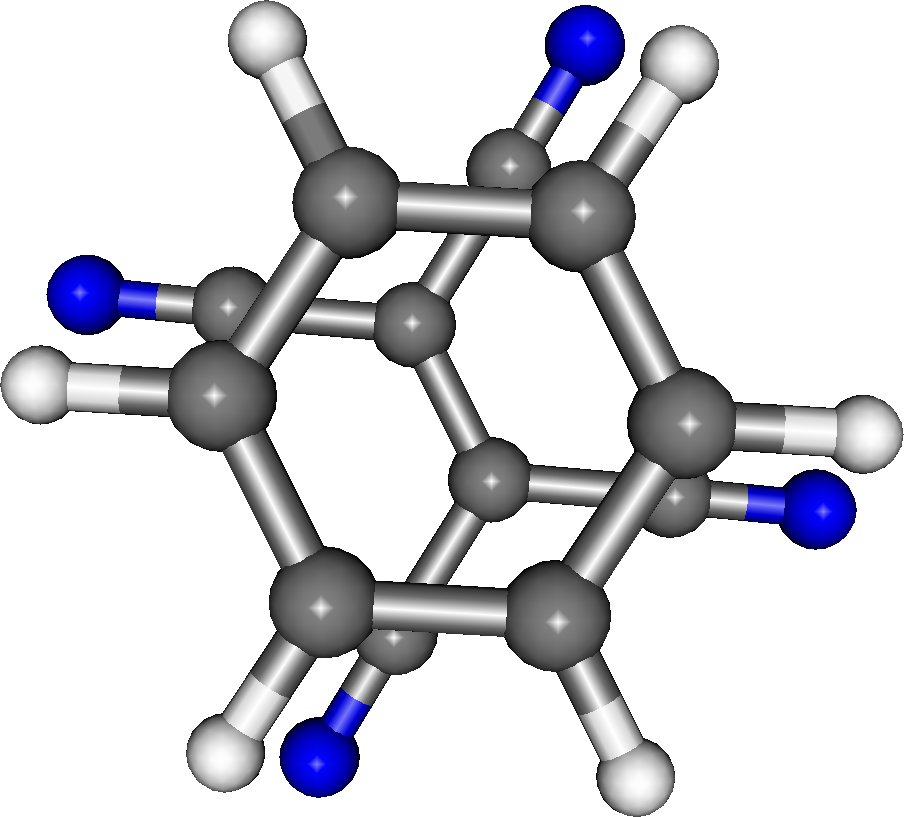
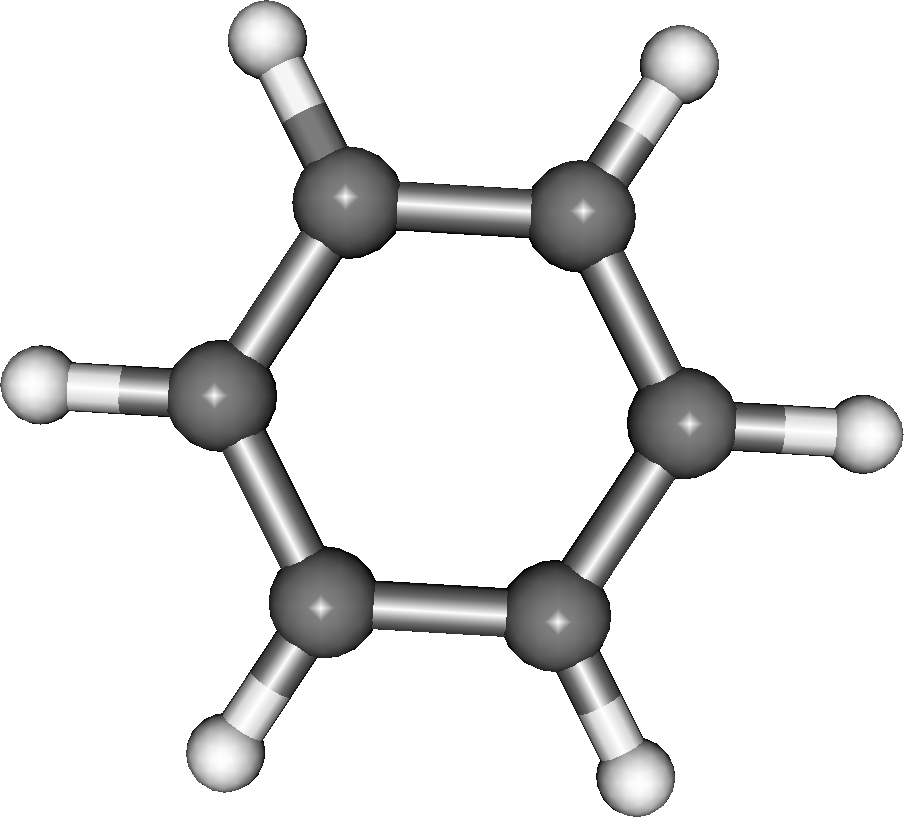
We will consider the benzene ring as the first fragment (fig. 1). It’s CLUSTER file is:
GGA-PBE*GGA-PBE
GRP
12
0.0000 2.6433 -3.9697 6 ALL
0.0000 -2.6433 -3.9697 6 ALL
2.2925 1.3220 -3.9702 6 ALL
-2.2925 1.3220 -3.9702 6 ALL
2.2925 -1.3220 -3.9702 6 ALL
-2.2925 -1.3220 -3.9702 6 ALL
0.0000 4.7079 -3.9776 1 ALL
0.0000 -4.7079 -3.9776 1 ALL
4.0794 2.3559 -3.9732 1 ALL
-4.0794 2.3559 -3.9732 1 ALL
-4.0794 -2.3559 -3.9732 1 ALL
4.0794 -2.3559 -3.9732 1 ALL
0.0000 0.0000
Once you run NRLMOL with this fragment, from EVALUES we get this information:
- Number of Bases: 324
- Number of occupied states: 21
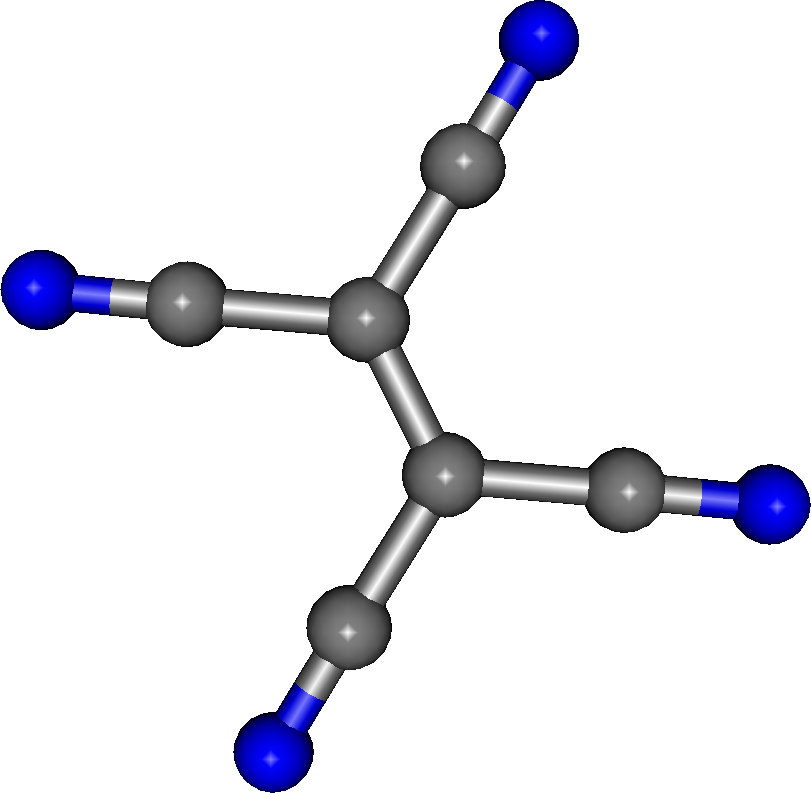
Now we run the second fragment (fig. 2), where the CLUSTER file is:
GGA-PBE*GGA-PBE
GRP
10
0.0000 1.2969 2.9150 6 ALL
0.0000 -1.2969 2.9150 6 ALL
-2.3072 2.7100 2.9357 6 ALL
2.3071 2.7100 2.9357 6 ALL
-2.3072 -2.7100 2.9357 6 ALL
2.3071 -2.7100 2.9357 6 ALL
-4.1665 3.8824 2.9754 7 ALL
4.1665 3.8824 2.9754 7 ALL
-4.1665 -3.8824 2.9754 7 ALL
4.1665 -3.8824 2.9754 7 ALL
0.0000 0.0000
The results we need from EVALUES are:
- Number of Bases: 350
- Number of occupied states: 32
Complex calculation¶
Now that we have run the fragments individually, must put them together and run NRLMOL on the complete complex (fig. 1). Here’s were you need the newest version of NRLMOL. The first thing to do is to set Y to FRAGMENTV as well as ‘SCF-ONLY’ in CALCTYPEV in NRLMOL_INPUT.DAT,your file should look something like this:
# Put Y,N or number next to the equal ...
# Don't forget thew quotation marks for the letters
# All variables in this list end with v
&input_data
ATOMSPHV = 'N'
CALCTYPEV = 'SCF-ONLY'
DFTD3V = 'N'
DIAG1V = 0
DIAG2V = 0
DOSOCCUV = 'N'
EXCITEDV = 'N'
FORMFAKV = 'N'
FRAGMENTV = 'Y'
JNTDOSV = 'N'
MATDIPOLEV = 'N'
NONSCFV = 'N'
NONSCFFORCESV = 'N'
WFGRIDV = 'N'
&end
The CLUSTER file is now a little different:
GGA-PBE*GGA-PBE
GRP
22 2
12 10
0.0000 2.6433 -3.9697 6 ALL
0.0000 -2.6433 -3.9697 6 ALL
2.2925 1.3220 -3.9702 6 ALL
-2.2925 1.3220 -3.9702 6 ALL
2.2925 -1.3220 -3.9702 6 ALL
-2.2925 -1.3220 -3.9702 6 ALL
0.0000 4.7079 -3.9776 1 ALL
0.0000 -4.7079 -3.9776 1 ALL
4.0794 2.3559 -3.9732 1 ALL
-4.0794 2.3559 -3.9732 1 ALL
-4.0794 -2.3559 -3.9732 1 ALL
4.0794 -2.3559 -3.9732 1 ALL
0.0000 1.2969 2.9150 6 ALL
0.0000 -1.2969 2.9150 6 ALL
-2.3072 2.7100 2.9357 6 ALL
2.3071 2.7100 2.9357 6 ALL
-2.3072 -2.7100 2.9357 6 ALL
2.3071 -2.7100 2.9357 6 ALL
-4.1665 3.8824 2.9754 7 ALL
4.1665 3.8824 2.9754 7 ALL
-4.1665 -3.8824 2.9754 7 ALL
4.1665 -3.8824 2.9754 7 ALL
0.0000 0.0000
The changes are:
- In line 3 we specify the total number of atoms (22)
- After that, in the same line, we specify the total number of fragments (2)
- Line 4 specifies the total number of atoms for fragment #1 (12), and
- The total number of atoms for fragment #2 (10)
By specifying the CLUSTER file this way, we are telling NRLMOL to generate the Hamiltonian matrix (written in Hamold) following this particular order of atoms and not group atoms by type, this was the matrix can be sliced according to the number of basis functions. Speaking of which when this CLUSTER file is run the number of bases from EVALUES is 674, which is the sum of the bases from the fragments. Also, the number of occupied states is 53. Again, a sum of the states of the individual fragments (we will need this value for the next step).
Fragment analysis¶
This is the actual fragment analysis, the two steps before this are only to setup this caclulation. This also invloves a different binary that must be constructed from the Mafile. On the command line construct this binary by issuing the command make fragment. This will create another binary called clusterfrag, this file runs serially so you can run it directly from the command line (however, it is still reccomended you use a script on external clusters). But before we start the calculation, some inputs are required: 1. create a new directory to run this binary. 2. put the following files there:
- The HAMOLD files for each of the fragments, renamed as HAM.1,HAM.2, etc.
- The HAMOLD file from the complex calculation
- The OVLBABY file from the complex calculation
- A new file called FRAGMENT.IN must be created, its structure is:
- Spin of the system (get the value of SPN from the first line in EVALUES)
- Number of fragments (2 for this case)
- Number of occupied states (53 for the complex)
- A listing of the number of bases and occupied states per fragment
- the following statement: .FALSE. which is used to indicate if it’s a restart of a calculation.
So the FRAGMENT.IN file should look like this:
1
2
53
324 21
350 32
.FALSE.
Once this binary runs, there should be two files created: OVERLAP and WAVEMO.